紡錘体チェックポイント
紡錘体チェックポイント(ぼうすいたいチェックポイント、英: spindle checkpoint)は、有糸分裂または減数分裂において、複製された各染色体が紡錘体に正しく接着する(後期)まで染色体の分離を防ぐ、細胞周期チェックポイントである。スピンドルチェックポイント、紡錘体(スピンドル)形成チェックポイント(英: spindle assembly checkpoint、略称: SAC)、有糸分裂チェックポイント(英: mitotic checkpoint)とも呼ばれる。適切な染色体分離を行うためには、姉妹染色分体上の2つのキネトコアはそれぞれ反対側の紡錘体極と接着していなければならない[1]。この接着パターンのときのみ、各娘細胞が染色体の1つのコピーを受け取ることが保証される。このチェックポイントの生化学的特徴はM期サイクリン/CDK複合体による後期促進複合体の刺激であり、これによってサイクリンや姉妹染色分体をつなぎとめているタンパク質の分解が引き起こされる[2]。

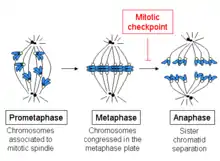
概要と重要性
中期の開始は、染色体のキネトコアへの微小管の連結と、細胞の中央部への染色体の整列によって特徴づけられる。各染色分体にはキネトコアが存在し、姉妹染色分体の各キネトコアに結合した微小管はそれぞれ細胞の反対側の極から発したものである。これらの微小管は染色体を細胞の両側の極へ引っ張る力を発生させるが、姉妹染色分体間の接着がこの力に対抗する。
中期から後期への移行段階では、この姉妹染色分体間の接着は解消され、分離された染色分体は紡錘体微小管によって細胞の両側の極へと引っ張られる。染色分体は紡錘体極の物理的な移動によってもさらに分離される。未成熟な段階で染色分体の分離が起こると、染色体の誤分離と娘細胞での染色体の異数性が引き起こされる可能性がある。そのため、中期でのチェックポイントは、染色体が適切に接着するまで後期への移行を防ぐ役割を果たす。
細胞の同一性と適切な機能を維持するためには、各細胞分裂後に適切な染色体の数が維持される必要がある。染色体数が正常よりも少ないまたは多い娘細胞が形成されるエラー(異数性と呼ばれる)が生じた場合、最善の場合には細胞死が引き起こされるが、壊滅的な表現型が生じる可能性もある[3][4]。
発見

Zirkleは、染色体の中期板への到着が1本でも遅れると、後期の開始はその到着の数分後まで引き伸ばされることを1970年に最初に観察した研究者の1人である[5]。この観察や同様の観察からは、中期から後期への移行段階には何らかの制御機構が存在することが示唆された。ノコダゾールやコルヒチンなどの薬剤を用いることで、紡錘体は解体され細胞周期は中期から後期への移行が防がれた[6]。推定される制御機構はSpindle Assembly Checkpoint(SAC)と名付けられ、以降精力的な研究が行われている[7]。
さまざまなタイプの遺伝学的研究により、紡錘体の脱重合[8][9]、2つのセントロメアを持つ染色体の存在[10]、セントロメアの異常な分離[11]、出芽酵母Saccharomyces cerevisiaeでの紡錘体極の欠陥[12]、キネトコアタンパク質の欠陥[13]、セントロメアDNAの変異[14]、有糸分裂で機能する分子モーターの欠陥[8]など、多様な種類の欠陥がSACを活性化することが明らかにされている[15]。
Zirkle[5]は自身の観察に基づき、細胞が後期へ進行するために必要な一部の物質がC(最後の染色体が中期板に到着するタイミング)の数分後、もしくはCと同時または直後の細胞質環境の劇的な変化の後に出現することを提唱し、この機能は紡錘体に接着していないキネトコア上で行われることを示唆した。McIntoshはこの提案を拡張し、張力に対して感受性のある酵素がセントロメアに位置しており、姉妹染色分体に双方の極からの張力が発生していないときには後期の開始の阻害因子を産生していることを示唆した[16]。事実、この「後期に入るのを待て」というシグナルはその大部分が未接着のキネトコア上またはそれに近接した領域で産生されていることをデータは示唆していた[17]。キネトコアの紡錘体への接着と関係した、阻害シグナルを不活性化し中期での停止を解除する主要なイベントは、キネトコアによる微小管の獲得[17]、または微小管のキネトコアへの固定を安定化する張力の発生[18]であった。その後、細胞質に2つの独立した紡錘体を持つ細胞での研究によって、中期から後期への移行の阻害因子は未接着のキネトコアによって産生され、それは細胞質を自由に拡散するものではないことが示された[19]。しかし同じ研究によって、細胞の一部でいったん中期から後期への移行が開始されると、この情報は細胞質全体に伝わり、未接着のキネトコアを含むもう一方の紡錘体による「後期に入るのを待て」というシグナルに打ち勝つことが示された。
姉妹染色分体の複製、接着、分離
細胞分裂: 複製と娘細胞への分配

細胞のサイズが十分に大きくなり、または適切な刺激を受けて細胞が分裂の準備が整った際には[20]、細胞は細胞周期の進行を開始する機構を活性化する。S期には中心体を含む大部分の細胞小器官の複製が行われる。そのため、細胞分裂過程が終結したときには、各娘細胞は完全な細胞小器官のセットを受け取ることとなる。それと同時に、S期にはDNAの複製が非常に正確に行われる必要がある。DNA複製が完了すると、真核生物ではDNA分子は凝縮され、分裂期染色体が形成される。各分裂期染色体は2つの姉妹染色分体から構成され、姉妹染色分体間の接着が確立されている。各染色分体は完全なDNA分子であり、細胞の2つの極にそれぞれ1つずつ位置する中心体のいずれかと微小管を介して接着される。中心体と微小管によって形成されるこの構造はその形状から紡錘体と呼ばれており、染色体は2つの中心体の間に保持される。姉妹染色分体間の接着は後期に切り離され、各染色分体は微小管を介して接着している中心体へ向かって移動する。このようにして、細胞分裂過程の終結時に娘細胞が切り離された際には、それぞれが完全な染色分体のセットを受け取る。細胞分裂時の姉妹染色分体の正確な分配を担う機構は染色体分離(chromosome segregation)と呼ばれている。
染色体分離が正しく行われることを保証するため、細胞は正確で複雑な機構を発達させている。まず、細胞はDNA複製と中心体の複製を協調的に行う必要があり、この過程に欠陥が生じると極が1つしかない、または多数の極を持つ紡錘体が形成される。こうした場合には染色体の分配のバランスが取れないため、異常な染色体分離が行われる[21]。
有糸分裂: 紡錘体への染色体の固定と染色体分離

S期の間に中心体は複製を行う。有糸分裂の開始時点で、双方の中心小体の長さは最大となり、さらなる物質をリクルートして微小管核形成能力を増大させる。有糸分裂が進行すると、双方の中心体は分離して紡錘体を形成する[22]。こうして紡錘体は微小管が発する2つの極を持つこととなる。微小管はタンパク質性の長い繊維で、非対称的な末端を持つ。一方の末端は(−)端と呼ばれ、比較的安定で中心体に近接している。もう一方の末端は(+)端と呼ばれ、伸長と短縮を繰り返しながら細胞の中心部で染色体を探索する。各染色分体にはセントロメアと呼ばれる特別な領域が存在し、その上にはキネトコアと呼ばれるタンパク質性の構造が組み立てられてられる。この構造は微小管の(+)を安定化することができる。そのため、細胞の中心部を探索している微小管が偶然にキネトコアと遭遇すると、キネトコアは微小管を捕捉し、染色体は姉妹染色分体の一方のキネトコアを介して紡錘体へ接着することとなる。染色体はキネトコアの紡錘体への接着に活発な役割を果たす。クロマチンにはRanのグアニンヌクレオチド交換因子(GEF)が結合しており、染色体近傍のRanはGDPの代わりにGTPの結合が促進される。活性化されたGTP結合型のRanは細胞質のタンパク質複合体からTPX2などの微小管安定化タンパク質を解離させ、染色体周辺で微小管の核形成と重合を誘導する[23]。こうしたキネトコア由来の微小管は、outer kinetochoreのキネシンモータータンパク質ともに、紡錘体由来の微小管の側面との相互作用を促進する。こうした側面との接着は不安定であり、末端型の接着へと変換される必要がある。側面型の接着から末端型の接着への変換によって、微小管の(+)端の伸長と短縮は、染色体の適切な二方向型(bi-orientation)の接着を達成するために染色体を押したり引いたりする力へと変換される。姉妹染色分体間は接着されており、また双方のキネトコアは双方の染色分体上で背中合わせに位置しているため、一方のキネトコアが1つの中心体に接着されると、もう一方のキネトコアは反対側の極に位置する中心体へ向かって露出するようになる。そのため、ほとんどの場合2つ目のキネトコアは微小管を介して反対側の極の中心体と結合し[24]、染色体は細胞分裂時の適切な分離が保証される基本的な二方向性配置(アンフィテリック(amphitelic)とも呼ばれる)となる[25][26]。時折、2つの姉妹キネトコアの1つが双方の極から発した微小管に同時に接着することがある。この配置はメロテリック(meroteric)と呼ばれ、紡錘体チェックポイントによっては検知されず、後期の間も中心部に取り残された染色体が形成され、異数性が生じる可能性がある。メロテリック型の配置は有糸分裂の初期には頻繁にみられるが、このタイプの配置はオーロラBによって検知されて取り除かれる[27]。オーロラBはさまざまなタイプの腫瘍で過剰発現しており、抗がん剤開発の標的となっている[28]。
コヒーシン: SMCタンパク質
上述したように、姉妹染色分体はS期(DNA複製によって2つの同一なコピー(染色分体)が生成される時期)から後期まで結合したままである。後期には、姉妹染色分体は分離され、分裂中の細胞のそれぞれ反対側の極へ向かって移動する。酵母とアフリカツメガエルXenopus laevis卵抽出液での遺伝学的・生化学的研究により、姉妹染色分体間の接着において必要不可欠な役割を果たしている、多数のタンパク質からなる複合体が同定されている[29]。この複合体はコヒーシン複合体として知られており、出芽酵母ではSmc1p、Smc3p、Scc1p(またはMcd1p)、Scc3pという少なくとも4つのサブユニットから構成されている。Smc1pとSmc3pは、高度に保存された染色体関連ATPアーゼのグループである、SMCタンパク質(Structural Maintenance of Chromosomes)と呼ばれるファミリーに属し、ヘテロ二量体(Smc1p/Smc3p)を形成する。Scc1pはRad21の出芽酵母ホモログであり、分裂酵母Schizosaccaromyces pombeでDNA修復に関与するタンパク質として最初に同定された。これら4つのタンパク質は酵母では必須であり、どれか1つにでも変異が生じると姉妹染色分体が早期に分離するようになる。酵母では、コヒーシンは染色体の腕に沿って優先的に結合し、セントロメアに近接して非常に豊富に存在することがクロマチン免疫沈降を用いた研究によって示されている[30]。
ヘテロクロマチンの役割
古典的な細胞学的観察から姉妹染色分体はヘテロクロマチン領域でより強固に接着していることが示唆されており[31]、ヘテロクロマチンの特別な構造または組成がコヒーシンのリクルートに有利に働いている可能性が示唆されている[32]。事実、分裂酵母ではSwi6(HP1の分裂酵母ホモログ)はヒストンH3のメチル化されたリジン9番残基に結合し、コヒーシンのセントロメアリピートへの結合を促進する[33][34]。より近年の研究からは、分裂酵母と脊椎動物の双方において、RNAi装置がヘテロクロマチンの確立を調節しており、この領域へのコヒーシンのリクルートを調節していることが示唆されている[35][36]。一方で、出芽酵母ではセントロメアに近接したヘテロクロマチン領域が存在しないにもかかわらず、機能的なセントロメアの存在によってそれに隣接する20–50 kbの領域でコヒーシンの結合の増加が誘導されることから、ヘテロクロマチン以外にもセントロメアでの接着の強化を保証する機構が存在するはずである[37]。
これに関連して、ヒト細胞では有糸分裂中にOrc2(S期のDNA複製の開始に関与する複製起点認識複合体(ORC)に含まれるタンパク質の1つ)もキネトコアに局在している[38]。一部の観察では酵母のOrc2は姉妹染色分体間の接着に関与することが示唆されており、その除去によってSACの活性化が誘導される[39]。また、ORC複合体の他の構成要素(分裂酵母のOrc5など)も接着に関与することが観察されている[40]。ORCタンパク質が関与する経路はコヒーシン経路に対して相加的に作用するようであるが、その大部分は未解明である。
接着の機能とその解消
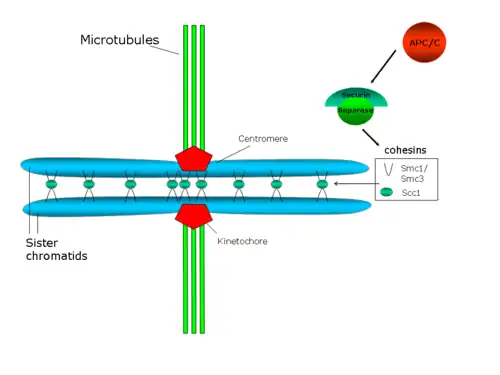
セントロメアでの接着は紡錘体微小管による極方向への力に抵抗し、姉妹キネトコア間に張力を発生させる。この張力は、オーロラBが関与すると考えられている機構によって、微小管-キネトコア間の接着を安定化する[41]。
事実、細胞内のコヒーシンレベルの低下は、姉妹染色分体の早期分離を引き起こすとともに、染色体の中期板への集合の欠陥や、オーロラBを含む染色体パッセンジャー複合体(chromosomal passenger complex)タンパク質の脱局在を引き起こす[42][43]。提唱されているコヒーシン複合体の構造からは、この複合体が双方の姉妹染色分体を直接的に連結していることが示唆されている[44]。この提唱構造では、コヒーシン複合体のSMC構成要素は構造的な役割を果たしているとされ、SMCヘテロ二量体はDNA結合タンパク質として機能し、そのコンフォメーションはATPによって調節されている可能性がある[45]。しかしながら、Scc1pとScc3pが調節的な役割を果たしている可能性もある[29]。
出芽酵母では、Pds1p(セキュリンとしても知られる)が姉妹染色分体間の接着を調節している。Pds1pはプロテアーゼEsp1p(セパラーゼまたはセパリン)に結合し、阻害を行う。後期の進行が開始されると、後期促進複合体(APC、APC/Cまたはサイクロソーム)がセキュリンを分解する。APC/CはRING型E3ユビキチンリガーゼであり、ユビキチンが付加されたE2ユビキチン結合酵素をリクルートする。セキュリンは、APC/Cの活性化サブユニットであるCdc20がAPC/Cのコアに結合している場合にのみ、APC/Cに認識される。セキュリン、Cdc20、E2酵素がすべてAPC/Cに結合すると、E2酵素はセキュリンをユビキチン化し、セキュリンは選択的に分解される。セキュリンの分解によってセパラーゼが放出され、セパラーゼは2つの姉妹染色分体を連結しているコヒーシンのリングを分解し、姉妹染色分体の分離が促進される[46]。Polo/Cdc5キナーゼがScc1の切断部位に隣接するセリン残基をリン酸化し、このリン酸化が切断活性を促進することも示されている[47]。
この装置は進化の過程を通じて保存されているが[48][49]、脊椎動物では大部分のコヒーシン分子はAPC/Cの存在とは無関係に前期に放出される。この過程はポロ様キナーゼPLK1とオーロラBに依存している[50]。しかし、ヒト細胞では少量のScc1は中期までセントロメアに結合したままであり、同程度の量が後期に切断されてセントロメアから消失する[51]。一方で一部の実験からは、姉妹染色分体の腕部の接着は姉妹セントロメアの分離後に徐々に失われることで、姉妹染色分体が細胞の反対側の極へ向かって移動することも示されている[52][53]。
染色体の腕部のコヒーシンの一部とセントロメアのコヒーシンはタンパク質シュゴシン(Sgo1)によって保護されることで、前期での放出を回避している[54][55]。Sgo1がセントロメアの接着の保護因子として機能するためには、Pds1と同様に後期の初めに不活性化される必要がある。事実、脊椎動物ではPds1とSgo1はどちらもAPC/Cの基質である[56]。
紡錘体チェックポイントの概要
紡錘体チェックポイント(SAC)は、不適切な接着がなされているキネトコアから発せられるシグナルであり、すべての真核生物の間で保存されている。SACはCdc20を負に制御することで細胞周期を停止し、それによって後期促進複合体(APC)のポリユビキチン化活性の活性化を防ぐ。SACシグナルを担うタンパク質は、MCC(mitotic checkpoint complex)と呼ばれる複合体を構成する。MCCには、SACタンパク質、Mad2/Mad3(BubR1)、Bub3、そしてCdc20が含まれる[57]。SACに関与する他のタンパク質には、Mad1、Bub1、Mps1、オーロラBがある。高等真核生物では、さらにROD-ZW10複合体の構成要素、p31comet、MAPK、CDK1-サイクリンB、Nek2、Plk1などの調節因子が存在する[58]。
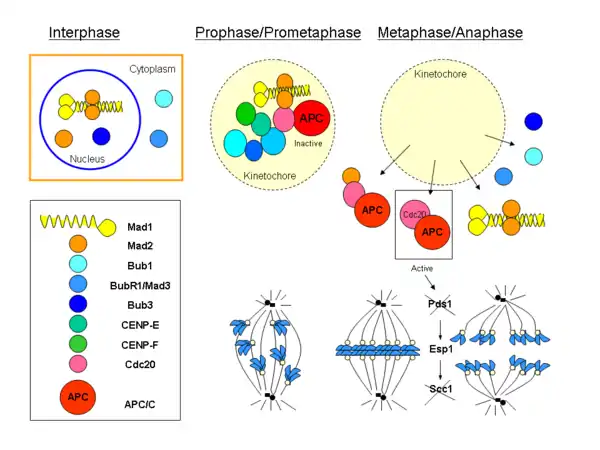
チェックポイントの活性化
SACは不適切に連結されたキネトコアと紡錘体微小管の間の相互作用を監視しており、キネトコアが紡錘体へ正しく接着されるまで維持される。前中期の間、Cdc20とSACタンパク質は紡錘体への接着の前にキネトコアへ濃縮される。これらのタンパク質は、除去されて正しいキネトコア-微小管間接着がなされるまで、SACの活性化状態を維持する。1つでも未接着のキネトコアが存在すると、SACは維持される[57]。微小管の(+)端の接着とキネトコア微小管の形成後、Mad1とMad2はキネトコアから除去される。チェックポイントの活性化の他の調節因子は、キネトコアの張力である。姉妹キネトコアがそれぞれ反対側の極に正しく接着しているときには、紡錘体の力によってキネトコアに張力が発生する。二方向型の接着がなされたキネトコアでの強い張力はキネトコアと微小管の組み立てを安定化するが、弱い張力は不安定化する作用がある。双方のキネトコアが一方の極に接着されるシンテリック(syntelic)型などの不適切な接着では、発生する張力の弱さによって不適切な接着は不安定化され、キネトコアが紡錘体へ正しい形で再接着を行うことが可能となる。染色体パッセンジャー複合体のオーロラB/Ipl1キナーゼが不適切なキネトコア接着の張力センサーとして機能し、微小管-キネトコア相互作用面に位置する、微小管切断KinI型キネシンMCAK、DASH複合体、Ndc80/Hec1複合体の制御を通じて不適切な接着を不安定化する[58][59]。オーロラB/Ipl1キナーゼは、1つのキネトコアが双方の極に同時に接着されているメロテリック(merotelic)型接着の修正にも重要である。メロテリック型接着は十分な張力を発生するため、SACによって検出されない。修正されない場合、染色分体のゆっくりとした移動速度のために染色体の誤分離が生じる可能性がある。
SACが活性化されると、MCCの活性の調節を介したAPCの阻害により、後期への移行は防がれる。MCCによるAPCの阻害機構はあまり解明されていないが、MCCはBubR1(Mad3)のKEN-boxモチーフを用いて偽基質としてAPCに結合すると考えられている。MCCの活性化と同時に、セントロメアタンパク質のCENP-EもBubR1を活性化し、後期の進行を防ぐ[58]。
MCCの形成
MCCはBub3と、Cdc20に結合したMad2、Mad3(BubR1)から構成される。Mad2とMad3はCdc20上の異なる部位に結合し、相乗的作用によってAPC/Cを阻害する。Bub3は、Mad3やBubR1のGLEBSモチーフと呼ばれるshort linear motifを介して結合を行う。MCCの形成のための結合の正確な順序はいまだ不明であるが、Mad2とCdc20が複合体を形成すると同時にBubR1、Bub3とCdc20も他の複合体を形成し、続いてこれら2つのサブ複合体が合体することでMCCが形成されている可能性がある[57]。ヒト細胞では、BubR1のCdc20への結合に先立ってMad2がCdc20に結合している必要があり、そのためMad-Cdc20サブ複合体がMCC形成の開始因子として機能している可能性がある。BubR1を欠失してもMad2-Cdc20のレベルはわずかに低下するのみである一方で、Mad2はBubR1-Bub3がCdc20に結合するために必要である。しかしながら、BubR1はチェックポイントの活性化には必要である[58]。
MCCの形成機構は明らかでなく、キネトコア依存的な形成と非依存的形成という競合する仮説が存在する。キネトコア非依存的形成を支持する証拠としては、キネトコアの組み立ての核となるタンパク質が変異した細胞やSACが不活性化された細胞でもMCCが検出されることが挙げられる。このことはMCCはキネトコアへの局在がなくとも有糸分裂期に組み立てられることを示唆している。あるモデルでは、前中期の未接着キネトコアは、機能的なSACを介してAPCをキネトコアへリクルートすることによって、APCのMCCによる阻害に対する感受性を高めているとされる。さらに、Mad2とBubR1の欠失はキネトコア非依存的に有糸分裂のタイミングに影響を与えるが、他のSACタンパク質の欠失は有糸分裂の持続期間に影響を与えることなくSACの機能不全を引き起こすことが明らかにされた。SACは、第一段階ではMad2とBubR1がキネトコア非依存的に有糸分裂の持続期間を制御し、第二段階では他のSACタンパク質と同様に未接着のキネトコアが存在する場合に延長を行うという、二段階のタイマーとして機能している可能性がある[58]。
一方で、現在主力となっているモデルは「Mad2鋳型モデル」であり、これはMCCの形成がMad2のキネトコアでのダイナミクスに依存するモデルである。Mad1は未接着のキネトコアに局在し、Mad2と強く結合する。Mad2とBubR1のキネトコアへの局在は、オーロラBにも依存している可能性がある[60]。オーロラBを持たない細胞は、染色体が微小管と接着していない場合でも中期での停止が起こらない[61]。Mad2には開いたコンフォメーション(O-Mad2)と閉じたコンフォメーション(C-Mad2)が存在する。未接着のキネトコアはまず Mad1/C-Mad2/p31comet複合体を結合し、未解明の機構によってp31cometを放出する。残ったMad1/C-Mad2複合体はO-Mad2をキネトコアへリクルートする。O-Mad2はコンフォメーション変化を起こし、C-Mad2となってMad1と結合する。このMad1/C-Mad2複合体はキネトコアへのさらなるO-Mad2のリクルートを担い、O-Mad2はC-Mad2へのコンフォメーション変化を起こしてCdc20に結合するという自己増幅反応が行われる。Mad1とCdc20はどちらも類似したMad2結合モチーフを持っているため、Cdc20への結合時にはC-Mad2へのコンフォメーション変化が起こる。このポジティブフィードバックループはp31cometによって負に調節されている。p31cometはMad1やCdc20に結合したC-Mad2に対して競合的に結合し、O-Mad2がC-Mad2へさらに結合していくことを防ぐ。下等真核生物にはp31cometが存在しないことを考えると、他の制御機構も存在する可能性がある。「鋳型モデル」という名称は、Mad1/C-Mad2がC-Mad2/Cdc20の形成の鋳型として機能することに由来している。こうしたC-Mad2/Cdc20複合体形成によるCdc20の隔離がSACの維持には必要不可欠である[57]。
チェックポイントの不活性化
姉妹染色分体の正しい二方向型接着がなされた後にSACを不活性化する機構はいくつか存在する。微小管-キネトコア間の接着に伴って、ダイニン複合体によるstripping(引きはがし)機構によってSACタンパク質はキネトコアから遠くへ輸送される[58]。引きはがされるタンパク質にはMad1、Mad2、Mps1、CENP-Fが含まれ、その後これらは紡錘体の極に再分布する。Strippingの過程は、未損傷の微小管構造と、微小管に沿ったダイニンの運動性に高度に依存している。p31cometはC-Mad2のポジティブフィードバックループの調節因子として機能するとともに、SACの不活性化因子としても作用する可能性がある。未接着のキネトコアは一時的にp31cometを不活性化するが、接着によって(おそらくリン酸化を介して)再活性化されてMad2の活性化を阻害する。他のSAC不活性化機構としては、Cdc20の非分解性ユビキチン化によるMad2-Cdc20複合体のエネルギー依存的な解離によるものがある。逆に、脱ユビキチン化酵素プロテクチンはSACの維持に必要である。未接着のキネトコアは継続的にMad2-Cdc20サブ複合体を再形成することによってチェックポイントを維持している。SACはAPCの活性化によるタンパク質分解によっても不活性化される可能性がある。サイクリンBのタンパク質分解とCDK1/サイクリンBの不活性化もSACの活性を阻害する。後期におけるMps1の分解は、姉妹染色分体間の接着の除去後のSACの再活性化を防ぐ。チェックポイントの不活性化後や細胞周期が正常に後期へ移行した場合、APCはMCCの活性の低下によって活性化される。このとき、酵素複合体は後期阻害因子であるセキュリンをポリユビキチン化する。セキュリンのユビキチン化と分解によって、セパラーゼと呼ばれるプロテアーゼが放出される。セパラーゼは姉妹染色分体を保持している接着分子を切断し、後期を活性化する[23]。
キネトコアでの末端型の微小管接着がSACシグナル伝達の特定の段階を破壊する過程の説明として、新たな機構も提唱されている。未接着のキネトコアでは、MCC形成の第一段階はキナーゼMps1によるSpc105のリン酸化である。その後、リン酸化されたSpc105は下流のシグナル伝達タンパク質である、Bub1、Bub3、Mad1、Mad2、Mad3、Cdc20をリクルートすることができるようになる。未接着キネトコアでのMad1との結合はMad2のコンフォメーション変化を引き起こし、O-Mad2からC-Mad2へ変換される。Mad1に結合したC-Mad2はその後、他のO-Mad2分子と二量体化し、Cdc20周辺でのC-Mad2への変換を触媒する。このC-Mad2/Cdc20複合体は、他のMCCが形成されるようキネトコアにMad1とC-Mad2を残して解離する。MCCはそれぞれ2分子のCdc20を隔離し、APC/Cとの相互作用を防ぐことによってSACを維持している[23]。Mps1によるSpc105のリン酸化はSACシグナル伝達経路の開始に必要かつ十分であるが、この段階はキネトコアへの微小管接着が存在しない場合にのみ起こる。内在性のMps1はNdc80のカルポニン相同ドメイン(CHドメイン)と相互作用することが示されており、Ndc80は染色体から離れたouter kinetochore領域に位置している。Mps1はouter kinetochoreにつながれているが、Ndc80の柔軟なヒンジ領域によってinner kinetochore内に局在しSpc105をリン酸化することができる。新たな提唱機構である機械的スイッチモデルでは、微小管のキネトコアへの末端型結合は2つの機構によってSACを不活性化するとされている。接着された微小管はNdc80のCHドメインとSpc105の間の距離を広げる。さらに、接着された微小管の周囲にリングを形成する、160のタンパク質からなる巨大複合体Dam1/DASH複合体が2つのタンパク質の間の障壁として作用する。この分離によってMps1とSpc105の間の相互作用が妨げられ、SACシグナル伝達経路は阻害される[62]。
このモデルは動物を含むより高等な生物でのSACの調節には当てはまらないことに留意しておくことは重要である。出芽酵母のキネトコア構造には1本の微小管しか接着しないが、動物のキネトコアは多数の微小管の結合部位を含むはるかに複雑な網目構造をしている[63]。そのため、SACの不活性化と後期への移行には、キネトコア結合部位のすべてに微小管が付着していることは必要とされない。すなわち、動物のキネトコアでは、SACが阻害されている間、微小管に接着した状態と接着していない状態が共存している。このモデルには、接着したキネトコアに結合したMps1が隣接する未接着キネトコアのSpc105をリン酸化するのを妨げるような障壁機構が含まれていない。さらに、酵母のDam1/DASH複合体に相当する複合体は動物細胞には見つかっていない。
SACの欠陥とがん
SACが適切に機能しない場合、染色体の誤分離や異数性、さらには腫瘍形成が生じる可能性がある[58]。形質転換はゲノムの完全性の維持が崩壊した時、特に染色体全体またはその大部分の領域で崩壊した時に生じ、加速される。事実、異数性はヒトの固形腫瘍の最も一般的な特徴であり、そのためSACは抗がん治療の標的となると考えられている[64]。通常、細胞周期のさまざまなチェックポイントは、高度に保存された冗長な機構を介してゲノムの完全性を管理しており、細胞の恒常性の維持や腫瘍形成の防止に重要な役割を果たしている。いくつかのSACタンパク質は、各細胞周期における適切な染色体分離を保証するために正と負の両方の調節因子として機能し、ゲノム不安定性とも呼ばれる染色体の不安定性を防いでいる。

ゲノムの完全性は現在いくつかのレベルで評価が行われており、一部の腫瘍では塩基置換、挿入、欠失などの不安定性がみられる一方、大部分の腫瘍では染色体全体の増加または喪失がみられる[65]。
有糸分裂調節タンパク質の変化が異数性を引き起こし、これががんでは高頻度で起こるという事実から[66]、当初はこれらの遺伝子ががん組織で変異している可能性があると考えられていた[67]。
一部のがんでは、形質転換を引き起こす欠陥の原因となる遺伝子はよく特徴づけられている。多発性骨髄腫などの血液のがんでは、イムノグロブリン遺伝子の再編成にDNA切断が必要であるという特有の性質のために、細胞遺伝学的な異常はきわめて一般的である。一方、多発性骨髄腫ではMAD2などの主にSACで機能するタンパク質の欠陥も特徴づけられている[68]。
また、大部分の固形腫瘍は主に異数体となっている。大腸がんに関しては、BUB1とBUBR1、そしてSTK15の増幅が、がんに至るゲノム不安定性への関与が示唆されている主要な調節因子である[69]。乳がんでは、BRCA1遺伝子によって特徴づけられる遺伝性のがんは散発性がんよりも高いレベルのゲノム不安定性を示す。BRCA1ヌル変異マウスは、重要なSACタンパク質であるMAD2の発現が低下することが実験的に示されている[70]。他のがんに関しては、異数性の原因を同定するためにはさらなる研究が必要である。
Mad2やBubR1などのタンパク質の生理的レベルの変化は明らかに異数性や腫瘍形成と関係しており、このことは動物モデルを用いて実証されている[71][72]。しかし、近年の研究ではそのシナリオより複雑なものであることが示されている。異数性は、組織でのSACの特定の構成要素のレベルの変化(低下または過剰発現のいずれか)によって腫瘍素因となる他の欠陥、すなわち、DNA損傷の増加、染色体再編成、細胞死の低下などが誘導されているときにのみ、高い腫瘍発生率をもたらす[73]。また、SACの一部の構成要素は有糸分裂外での機能、Mad1は核内移行、Bub3は転写抑制、BubR1は細胞死、DNA損傷応答、老化、巨核球産生への関与が示唆されている。このことはすべて、腫瘍形成の増加が異数性だけではない他の欠陥とも関連していることを支持するものである[73]。
BUB1やBUBR1のようにチェックポイントに影響を与えることが知られているがん関連変異は、実際のところは稀である。しかしながら、がんへの関与が示唆されているいくつかのタンパク質には紡錘体形成ネットワークとの関わりが存在する。p53などの主要ながん抑制因子もSACに役割を果たしている。ヒトのがんで最も一般的に変異している遺伝子であるp53が存在しない場合、細胞周期チェックポイントには大きな影響が生じる。p53はG1期チェックポイントで作用することが示されていたが、SACの調節にも同様に重要であるようである[74]。また、がんの重要な面の1つとして、細胞死またはアポトーシスの阻害が挙げられる。IAP(inhibitor of apoptosis)ファミリーのメンバーであるサバイビンは、中心体近傍の紡錘体微小管と中期染色体のキネトコアに局在している。サバイビンはアポトーシスを阻害して腫瘍形成を促進するだけでなく、染色体分離や、より原初的な生物での役割と同様に有糸分裂の終盤段階の重要な調節因子であることがノックアウトマウスを用いた研究により示唆されている[75]。
キネトコアの接着、微小管の機能、姉妹染色分体間の接着などのSACの他の側面も、欠陥が生じて異数性が引き起こされる可能性がある。がん細胞は、SACの回避によって多方向に分裂し、多極型の有糸分裂を引き起こすことが観察されている[76]。多極型紡錘体での中期から後期への移行は不完全なセパラーゼサイクルを介して行われ、結果として染色体不分離が高頻度で生じ、がん細胞の異数性を増幅させる。
SACを標的としたがん治療
この研究分野の進展によって、紡錘体形成の欠陥を標的とした治療法の開発が始まっている。ビンカアルカロイドやタキサンなどの古い治療法は紡錘体形成に加わる微小管を標的とし、微小管のダイナミクスを破壊することでSACによって細胞周期を停止させ、最終的に細胞死を引き起こすものである[77]。パクリタキセルとドセタキセルはどちらも現在でも乳がん、卵巣がんや他の上皮性がんの治療に用いられている。しかし、こうした治療法は多くの場合、副作用や薬剤抵抗性が高いという特徴がある。
SACに影響を与える調節因子ネットワーク内の他の標的の探索も行われており、オーロラキナーゼタンパク質に高い関心が寄せられている[78]。オーロラAは増幅された場合、SACを無効化するがん遺伝子として作用し、正常でない後期の開始と異数性、そしてパクリタキセルに対する抵抗性も示す[79]。オーロラAの低分子阻害剤はin vivoモデルでの抗腫瘍効果が示されており、さらなる臨床開発の良い標的である可能性が示唆される[80]。オーロラBの阻害剤も臨床開発が行われており、異常なキネトコア-微小管間接着をもたらし、同様にチェックポイントを無効化する[78]。サバイビンも複数の経路の主要なノードとして機能するため臨床的な治療法開発の魅力的な分子標的であり、紡錘体形成とチェックポイントの制御もサバイビンが関与する経路の1つである[81]。さらに、KSPのような有糸分裂関連モータータンパク質の阻害も検討されている。近年臨床試験が開始されたこれらの阻害剤は、SACによって有糸分裂の停止を引き起こし、アポトーシスを誘導する[3][82]。
出典
- “The life and miracles of kinetochores”. The EMBO Journal 28 (17): 2511–31. (September 2009). doi:10.1038/emboj.2009.173. PMC 2722247. PMID 19629042.
- Morgan, David Owen, 1958- (2007). The cell cycle : principles of control. London: New Science Press. ISBN 978-0-19-920610-0. OCLC 70173205
- Sinha, Debottam; Duijf, Pascal H. G.; Khanna, Kum Kum (01 2019). “Mitotic slippage: an old tale with a new twist”. Cell Cycle (Georgetown, Tex.) 18 (1): 7–15. doi:10.1080/15384101.2018.1559557. ISSN 1551-4005. PMC 6343733. PMID 30601084.
- “Short- and long-term effects of chromosome mis-segregation and aneuploidy”. Nature Reviews Molecular Cell Biology 16 (8): 473–85. (August 2015). doi:10.1038/nrm4025. hdl:1721.1/117201. PMID 26204159.
- “Ultraviolet-microbeam irradiation of newt-cell cytoplasm: spindle destruction, false anaphase, and delay of true anaphase”. Radiation Research 41 (3): 516–37. (March 1970). Bibcode: 1970RadR...41..516Z. doi:10.2307/3572841. JSTOR 3572841. PMID 5438206.
- “Colcemid and the mitotic cycle”. Journal of Cell Science 102 ( Pt 3) (3): 387–92. (July 1992). PMID 1506421.
- “Linking kinetochore-microtubule binding to the spindle checkpoint”. Developmental Cell 14 (4): 474–9. (April 2008). doi:10.1016/j.devcel.2008.03.015. PMC 2696048. PMID 18410725.
- “Feedback control of mitosis in budding yeast”. Cell 66 (3): 519–31. (August 1991). doi:10.1016/0092-8674(81)90015-5. PMID 1651172.
- “S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function”. Cell 66 (3): 507–17. (August 1991). doi:10.1016/0092-8674(81)90014-3. PMID 1651171.
- “A delay in the Saccharomyces cerevisiae cell cycle that is induced by a dicentric chromosome and dependent upon mitotic checkpoints”. Molecular and Cellular Biology 12 (9): 3857–64. (September 1992). doi:10.1128/MCB.12.9.3857. PMC 360258. PMID 1324407.
- “Aberrantly segregating centromeres activate the spindle assembly checkpoint in budding yeast”. The Journal of Cell Biology 133 (1): 75–84. (April 1996). doi:10.1083/jcb.133.1.75. PMC 2120768. PMID 8601615.
- “Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption”. Science 273 (5277): 953–6. (August 1996). Bibcode: 1996Sci...273..953H. doi:10.1126/science.273.5277.953. PMID 8688079.
- “Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae”. Molecular and Cellular Biology 15 (12): 6838–44. (December 1995). doi:10.1128/MCB.15.12.6838. PMC 230938. PMID 8524250.
- “Centromere DNA mutations induce a mitotic delay in Saccharomyces cerevisiae”. Proceedings of the National Academy of Sciences of the United States of America 89 (19): 8908–12. (October 1992). Bibcode: 1992PNAS...89.8908S. doi:10.1073/pnas.89.19.8908. JSTOR 2360300. PMC 50033. PMID 1409584.
- “Lesions in many different spindle components activate the spindle checkpoint in the budding yeast Saccharomyces cerevisiae”. Genetics 152 (2): 509–18. (June 1999). PMC 1460633. PMID 10353895.
- “Structural and mechanical control of mitotic progression”. Cold Spring Harbor Symposia on Quantitative Biology 56: 613–9. (1991). doi:10.1101/sqb.1991.056.01.070. PMID 1819511.
- “The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores”. The Journal of Cell Biology 130 (4): 941–8. (August 1995). doi:10.1083/jcb.130.4.941. PMC 2199954. PMID 7642709.
- “Tension-sensitive kinetochore phosphorylation and the chromosome distribution checkpoint in praying mantid spermatocytes”. Journal of Cell Science 110 ( Pt 5) (5): 537–45. (March 1997). PMID 9092936.
- “Mitosis in vertebrate somatic cells with two spindles: implications for the metaphase/anaphase transition checkpoint and cleavage”. Proceedings of the National Academy of Sciences of the United States of America 94 (10): 5107–12. (May 1997). Bibcode: 1997PNAS...94.5107R. doi:10.1073/pnas.94.10.5107. PMC 24639. PMID 9144198.
- “Size control in animal development”. Cell 96 (2): 235–44. (January 1999). doi:10.1016/S0092-8674(00)80563-2. PMID 9988218.
- “Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A”. Nature Cell Biology 1 (2): 88–93. (June 1999). doi:10.1038/10054. PMID 10559879.
- “Protein kinases in control of the centrosome cycle”. FEBS Letters 452 (1–2): 92–5. (June 1999). doi:10.1016/S0014-5793(99)00534-7. PMID 10376685.
- Morgan, David O. (2006-09-06). The Cell Cycle: Principles of Control (Primers in Biology) (1 ed.). New Science Press, Ltd. ISBN 978-0-87893-508-6
- “How cells get the right chromosomes”. Science 275 (5300): 632–7. (January 1997). doi:10.1126/science.275.5300.632. PMID 9005842.
- “The centromere geometry essential for keeping mitosis error free is controlled by spindle forces”. Nature 450 (7170): 745–9. (November 2007). Bibcode: 2007Natur.450..745L. doi:10.1038/nature06344. PMC 2586812. PMID 18046416.
- “Tension between two kinetochores suffices for their bi-orientation on the mitotic spindle”. Nature 428 (6978): 93–7. (March 2004). Bibcode: 2004Natur.428...93D. doi:10.1038/nature02328. PMID 14961024.
- “Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors”. Current Biology 16 (17): 1711–8. (September 2006). doi:10.1016/j.cub.2006.07.022. PMID 16950108.
- “Aurora kinases as anticancer drug targets”. Clinical Cancer Research 14 (6): 1639–48. (March 2008). doi:10.1158/1078-0432.CCR-07-2179. PMID 18347165.
- “Chromosome cohesion, condensation, and separation”. Annual Review of Biochemistry 69: 115–44. (2000). doi:10.1146/annurev.biochem.69.1.115. PMID 10966455.
- “Establishment and maintenance of sister chromatid cohesion in fission yeast by a unique mechanism”. The EMBO Journal 20 (20): 5779–90. (October 2001). doi:10.1093/emboj/20.20.5779. PMC 125673. PMID 11598020.
- “The spindle is required for the process of sister chromatid separation in Drosophila neuroblasts”. Experimental Cell Research 192 (1): 10–5. (January 1991). doi:10.1016/0014-4827(91)90150-S. PMID 1898588.
- “Shaping the metaphase chromosome: coordination of cohesion and condensation”. BioEssays 23 (10): 924–35. (October 2001). doi:10.1002/bies.1133. PMID 11598959.
- “Requirement of heterochromatin for cohesion at centromeres”. Science 294 (5551): 2539–42. (December 2001). Bibcode: 2001Sci...294.2539B. doi:10.1126/science.1064027. PMID 11598266.
- “Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast”. Nature Cell Biology 4 (1): 89–93. (January 2002). doi:10.1038/ncb739. PMID 11780129.
- “RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast”. Proceedings of the National Academy of Sciences of the United States of America 100 (1): 193–8. (January 2003). Bibcode: 2003PNAS..100..193H. doi:10.1073/pnas.232688099. PMC 140924. PMID 12509501.
- “Dicer is essential for formation of the heterochromatin structure in vertebrate cells”. Nature Cell Biology 6 (8): 784–91. (August 2004). doi:10.1038/ncb1155. PMID 15247924.
- “The kinetochore is an enhancer of pericentric cohesin binding”. PLOS Biology 2 (9): E260. (September 2004). doi:10.1371/journal.pbio.0020260. PMC 490027. PMID 15309047.
- “Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance”. The EMBO Journal 23 (13): 2651–63. (July 2004). doi:10.1038/sj.emboj.7600255. PMC 449767. PMID 15215892.
- “The origin recognition complex functions in sister-chromatid cohesion in Saccharomyces cerevisiae”. Cell 128 (1): 85–99. (January 2007). doi:10.1016/j.cell.2006.11.045. PMID 17218257.
- “Schizosaccharomyces pombe Orc5 plays multiple roles in the maintenance of genome stability throughout the cell cycle”. Cell Cycle 7 (8): 1085–96. (April 2008). doi:10.4161/cc.7.8.5710. PMID 18414064.
- “Kinetochore orientation in mitosis and meiosis”. Cell 119 (3): 317–27. (October 2004). doi:10.1016/j.cell.2004.10.014. PMID 15507205.
- “Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells”. Developmental Cell 1 (6): 759–70. (December 2001). doi:10.1016/S1534-5807(01)00088-0. PMID 11740938.
- “Depletion of Drad21/Scc1 in Drosophila cells leads to instability of the cohesin complex and disruption of mitotic progression”. Current Biology 13 (3): 208–18. (February 2003). doi:10.1016/S0960-9822(03)00047-2. PMID 12573216.
- “Molecular architecture of SMC proteins and the yeast cohesin complex”. Molecular Cell 9 (4): 773–88. (April 2002). doi:10.1016/S1097-2765(02)00515-4. PMID 11983169.
- “SMC-mediated chromosome mechanics: a conserved scheme from bacteria to vertebrates?”. Genes & Development 13 (1): 11–9. (January 1999). doi:10.1101/gad.13.1.11. PMID 9887095.
- “An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast”. Cell 93 (6): 1067–76. (June 1998). doi:10.1016/S0092-8674(00)81211-8. PMID 9635435.
- “Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast”. Cell 105 (4): 459–72. (May 2001). doi:10.1016/S0092-8674(01)00362-2. PMID 11371343.
- “Degradation of Drosophila PIM regulates sister chromatid separation during mitosis”. Genes & Development 14 (17): 2192–205. (September 2000). doi:10.1101/gad.176700. PMC 316890. PMID 10970883.
- “Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis”. The EMBO Journal 20 (4): 792–801. (February 2001). doi:10.1093/emboj/20.4.792. PMC 145417. PMID 11179223.
- “Characterization of vertebrate cohesin complexes and their regulation in prophase”. The Journal of Cell Biology 151 (4): 749–62. (November 2000). doi:10.1083/jcb.151.4.749. PMC 2169443. PMID 11076961.
- “Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes”. The Journal of Cell Biology 150 (3): 405–16. (August 2000). doi:10.1083/jcb.150.3.405. PMC 2175199. PMID 10931856.
- “Regulation of sister chromatid cohesion between chromosome arms”. Current Biology 14 (13): 1187–93. (July 2004). doi:10.1016/j.cub.2004.06.052. PMID 15242616.
- “Micromanipulation of chromosomes reveals that cohesion release during cell division is gradual and does not require tension”. Current Biology 14 (23): 2124–9. (December 2004). doi:10.1016/j.cub.2004.11.052. PMID 15589155.
- “The complete removal of cohesin from chromosome arms depends on separase”. Journal of Cell Science 120 (Pt 23): 4188–96. (December 2007). doi:10.1242/jcs.011528. PMID 18003702.
- “Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells”. PLOS Biology 3 (3): e86. (March 2005). doi:10.1371/journal.pbio.0030086. PMC 1054882. PMID 15737064.
- “Vertebrate shugoshin links sister centromere cohesion and kinetochore microtubule stability in mitosis”. Cell 118 (5): 567–78. (September 2004). doi:10.1016/j.cell.2004.08.016. PMID 15339662.
- “The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint”. Current Biology 15 (3): 214–25. (February 2005). doi:10.1016/j.cub.2005.01.038. PMID 15694304.
- “The spindle-assembly checkpoint in space and time”. Nature Reviews. Molecular Cell Biology 8 (5): 379–93. (May 2007). doi:10.1038/nrm2163. PMID 17426725.
- “Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2”. Science 297 (5590): 2267–70. (September 2002). Bibcode: 2002Sci...297.2267M. doi:10.1126/science.1075596. PMID 12351790.
- “Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension”. The EMBO Journal 22 (12): 2934–47. (June 2003). doi:10.1093/emboj/cdg307. PMC 162159. PMID 12805209.
- “The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint”. The Journal of Cell Biology 161 (2): 281–94. (April 2003). doi:10.1083/jcb.200208092. PMC 2172906. PMID 12707311.
- “The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling”. Nature Cell Biology 17 (7): 868–79. (July 2015). doi:10.1038/ncb3179. PMC 4630029. PMID 26053220.
- Molecular Biology of The Cell (6th ed.). New York, NY: Garland Science, Taylor & Francis Group. (2015). pp. 988. ISBN 978-0-8153-4432-2
- “On the road to cancer: aneuploidy and the mitotic checkpoint”. Nature Reviews. Cancer 5 (10): 773–85. (October 2005). doi:10.1038/nrc1714. PMID 16195750.
- “Genetic instabilities in human cancers”. Nature 396 (6712): 643–9. (December 1998). Bibcode: 1998Natur.396..643L. doi:10.1038/25292. PMID 9872311.
- “Does aneuploidy cause cancer?”. Current Opinion in Cell Biology 18 (6): 658–67. (December 2006). doi:10.1016/j.ceb.2006.10.002. PMID 17046232.
- “Mutations of mitotic checkpoint genes in human cancers”. Nature 392 (6673): 300–3. (March 1998). Bibcode: 1998Natur.392..300C. doi:10.1038/32688. PMID 9521327.
- Díaz-Rodríguez, Elena; Álvarez-Fernández, Stela; Chen, Xi; Paiva, Bruno; López-Pérez, Ricardo; García-Hernández, Juan Luis; San Miguel, Jesús F.; Pandiella, Atanasio (2011). “Deficient spindle assembly checkpoint in multiple myeloma”. PloS One 6 (11): e27583. doi:10.1371/journal.pone.0027583. ISSN 1932-6203. PMC 3223182. PMID 22132115.
- Grady, William M. (2004). “Genomic instability and colon cancer”. Cancer and Metastasis Reviews 23 (1–2): 11–27. doi:10.1023/A:1025861527711. PMID 15000146.
- “A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint”. Proceedings of the National Academy of Sciences of the United States of America 101 (49): 17108–13. (December 2004). Bibcode: 2004PNAS..10117108W. doi:10.1073/pnas.0407585101. PMC 535394. PMID 15563594.
- “Mad2 overexpression promotes aneuploidy and tumorigenesis in mice”. Cancer Cell 11 (1): 9–23. (January 2007). doi:10.1016/j.ccr.2006.10.019. PMC 1850996. PMID 17189715.
- “Overexpression of BUBR1 is associated with chromosomal instability in bladder cancer”. Cancer Genetics and Cytogenetics 174 (1): 42–7. (April 2007). doi:10.1016/j.cancergencyto.2006.11.012. PMID 17350465.
- “The role of aneuploidy in promoting and suppressing tumors”. The Journal of Cell Biology 185 (6): 935–7. (June 2009). doi:10.1083/jcb.200905098. PMC 2711620. PMID 19528293.
- Cross, Shawn M.; Sanchez, Carissa A; Morgan, Catherine A.; Schimke, Melana K.; Reid, Brian J. (1995). “A p53-dependant mouse spindle checkpoint”. Science 3 (5202): 1353–1356. Bibcode: 1995Sci...267.1353C. doi:10.1126/science.7871434. PMID 7871434.
- “The molecular basis and potential role of survivin in cancer diagnosis and therapy”. Trends in Molecular Medicine 7 (12): 542–7. (December 2001). doi:10.1016/S1471-4914(01)02243-2. PMID 11733216.
- Gisselsson, David; Håkanson, Ulf; Stoller, Patrick; Marti, Dominik; Jin, Yuesheng; Rosengren, Anders H.; Stewénius, Ylva; Kahl, Fredrik et al. (2008-04-02). “When the genome plays dice: circumvention of the spindle assembly checkpoint and near-random chromosome segregation in multipolar cancer cell mitoses”. PloS One 3 (4): e1871. doi:10.1371/journal.pone.0001871. ISSN 1932-6203. PMC 2289843. PMID 18392149.
- “Targeting microtubules for cancer chemotherapy”. Current Medicinal Chemistry. Anti-Cancer Agents 5 (1): 65–71. (January 2005). doi:10.2174/1568011053352569. PMID 15720262.
- “Aurora kinases: new targets for cancer therapy”. Clinical Cancer Research 12 (23): 6869–75. (December 2006). doi:10.1158/1078-0432.CCR-06-1405. PMID 17145803.
- “AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol”. Cancer Cell 3 (1): 51–62. (January 2003). doi:10.1016/S1535-6108(02)00235-0. PMID 12559175.
- “VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo”. Nature Medicine 10 (3): 262–7. (March 2004). doi:10.1038/nm1003. PMID 14981513.
- “Survivin, cancer networks and pathway-directed drug discovery”. Nature Reviews. Cancer 8 (1): 61–70. (January 2008). doi:10.1038/nrc2293. PMID 18075512.
- “Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage”. Cancer Cell 8 (1): 49–59. (July 2005). doi:10.1016/j.ccr.2005.06.003. PMID 16023598.
関連文献
- “Structural analysis of Bub3 interactions in the mitotic spindle checkpoint”. Proceedings of the National Academy of Sciences of the United States of America 104 (4): 1201–6. (January 2007). Bibcode: 2007PNAS..104.1201L. doi:10.1073/pnas.0610358104. PMC 1770893. PMID 17227844.
- “The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins”. The Journal of Biological Chemistry 276 (28): 26559–67. (July 2001). doi:10.1074/jbc.M101083200. PMID 11352911.
- “Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans”. Nature Cell Biology 1 (8): 514–21. (December 1999). doi:10.1038/70309. PMID 10587648.